
Ein strategischer Leitfaden für Produktmanager, CTOs und Medizinproduktehersteller
Medical Design, das Regulatorik, Usability und Innovation verbindet
Was ist Medical Design?
Medical Design ist die Disziplin der Entwicklung von Medizinprodukten, die sicher, wirksam und gebrauchstauglich sind. Anders als im Consumer-Produktdesign muss jede Entscheidung einem regulatorischen Rahmen genügen: MDR 2017/745/EU für den europäischen Marktzugang, IEC 62366 für Usability Engineering, ISO 14971 für Risikomanagement und ISO 13485 für Qualitätsmanagementsysteme.
Doch Konformität allein macht noch kein gutes Produkt. Das beste Medical Design bringt regulatorische Anforderungen mit klinischen Workflows, Nutzerempathie und Fertigbarkeit in Einklang. Hier treffen Engineering und Human Factors aufeinander — wo das Gewicht eines chirurgischen Instruments, das Feedback eines Beatmungsgeräte-Buttons und die Lesbarkeit eines Infusionspumpen-Displays klinische Konsequenzen haben.
Für Produktmanager und CTOs ist Medical Design keine nachgelagerte Styling-Übung. Es ist eine strategische Disziplin, die über Time-to-Market, CE-Kennzeichnung, Nutzerakzeptanz und langfristige Patientensicherheit entscheidet.
Warum ist Medical Design geschäftsentscheidend?

Die Zahlen sprechen eine klare Sprache. Der globale Medizinproduktemarkt erreichte 2025 ein Volumen von 586 Milliarden US-Dollar und soll bis 2035 auf 1.084 Milliarden wachsen — bei einem CAGR von 6,34 % (Quelle: Towards Healthcare). Allein das Segment für Design- und Entwicklungsdienstleistungen wird auf 13,29 Milliarden geschätzt und wächst bis 2034 auf 28,08 Milliarden bei 8,6 % CAGR (Quelle: Fortune Business Insights).
Tragbare Medizinprodukte werden 2026 die 60-Milliarden-Dollar-Marke überschreiten. Schlüsseltrends für 2026: KI-gestützte Diagnostik, Point-of-Care-Tests, nachhaltige Materialien und Roboterchirurgie. Die EUDAMED-Datenbank wird im Mai 2026 verpflichtend — eine weitere Compliance-Ebene.
Laut Boehms Kosteneskaltionsmodell kosten späte Usability-Probleme 10- bis 100-mal mehr als Fehler, die in frühen formativen Evaluationen erkannt werden. Für Produktmanager ist Medical Design kein Kostenfaktor — es ist der kritische Pfad zu Marktzugang, Wettbewerbsdifferenzierung und Patientensicherheit.
Welcher regulatorische Rahmen gilt für Medical Design in Europa?
Drei Säulen definieren die regulatorische Landschaft für das Medical Design in Europa. Sie zu kennen ist keine Option — es ist die Voraussetzung für die CE-Kennzeichnung.
Was fordert die MDR 2017/745/EU von Herstellern?
Die Medizinprodukteverordnung (MDR 2017/745/EU) hat die ältere Medizinprodukterichtlinie (MDD) im Mai 2021 abgelöst. Sie führte strengere Klassifizierungsregeln, eine verpflichtende klinische Bewertung für alle Risikoklassen, umfassende technische Dokumentation und kontinuierliche Post-Market-Surveillance ein. Die Verordnung trifft kleine und mittlere Hersteller besonders hart — viele haben Produkteinführungen verschoben oder den EU-Markt ganz verlassen. Ein Reformvorschlag vom Dezember 2025 zielt darauf ab, die Belastung für KMU zu verringern, ohne die Sicherheitsstandards aufzugeben. Für Designer bedeutet MDR: Jede Materialwahl, jede Interface-Entscheidung und jedes Beschriftungsdetail muss dokumentiert und begründet werden.
Wie strukturiert die IEC 62366 den Usability-Engineering-Prozess?
Die IEC 62366 ist die internationale Norm, die festlegt, wie Hersteller Usability Engineering in die Medizinprodukteentwicklung integrieren müssen. Sie fordert Nutzungskontextanalyse, Benutzerschnittstellenspezifikation, Identifikation nutzungsbezogener Gefährdungen sowie formative und summative Evaluation. Anwendungsfehler sind die häufigste Einzelursache für Medizinprodukte-Vorfälle — die IEC 62366 existiert, um sie zu verhindern. Benannte Stellen erwarten dokumentierte Nachweise der IEC-62366-Konformität für die CE-Kennzeichnung. Die Norm gilt für jedes Gerät, von der einfachen Blutdruckmanschette bis zum komplexen chirurgischen Roboter. Für Medical-Design-Teams ist sie kein bürokratischer Aufwand — sie ist das Rahmenwerk, das Patienten schützt.
Warum ist Risikomanagement nach ISO 14971 designrelevant?
Die ISO 14971 fordert systematische Identifikation, Einschätzung und Kontrolle von Risiken über den gesamten Produktlebenszyklus. Für Designer bedeutet das: Jede Entscheidung hat eine Risikodimension — Materialauswahl, Interface-Layout, physische Ergonomie, Reinigungsverfahren — alles fließt in die Risikomanagementakte ein. Usability Engineering nach IEC 62366 und Risikomanagement nach ISO 14971 sind zwei Seiten derselben Medaille. Ein Griff, der in einem nassen Operationssaal zu glatt zum sicheren Halten ist, ist nicht nur ein Designfehler — es ist ein Risiko, das identifiziert, bewertet und kontrolliert werden muss.

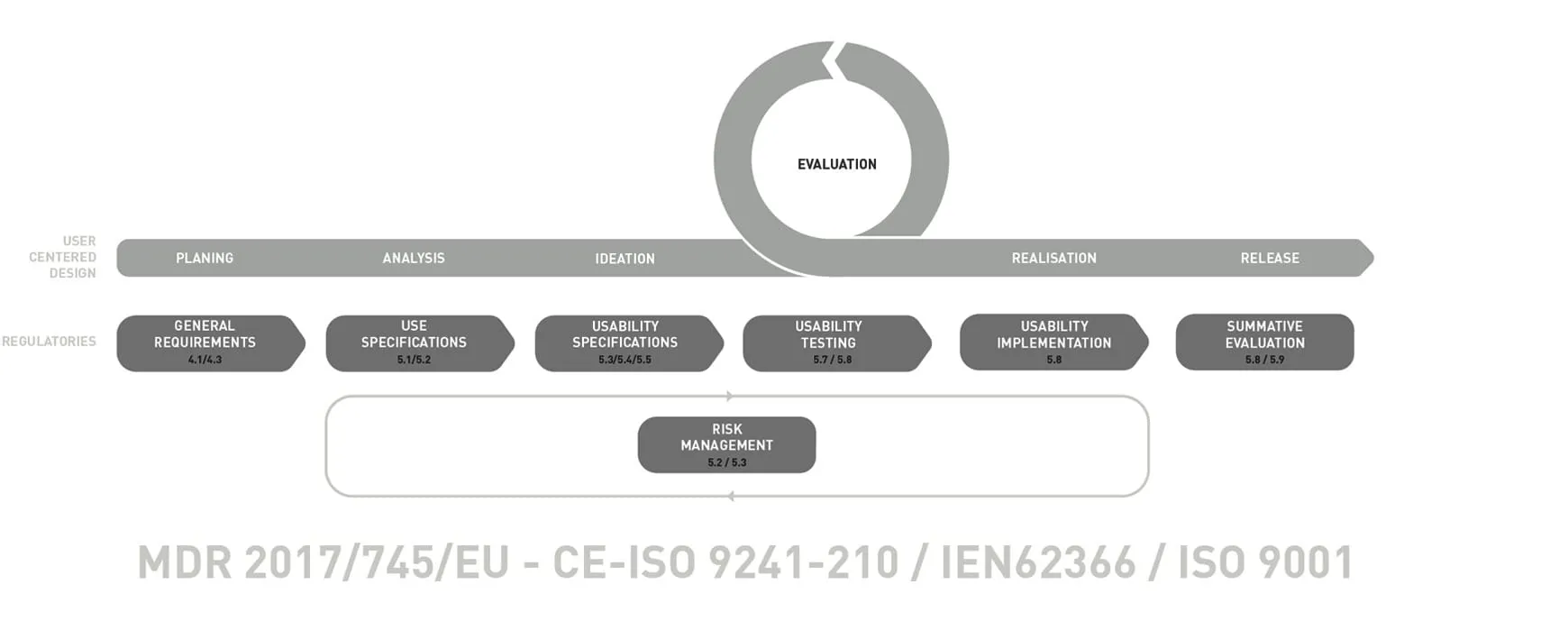
Wie funktioniert ein siebenstufiger Usability-Engineering-Prozess?
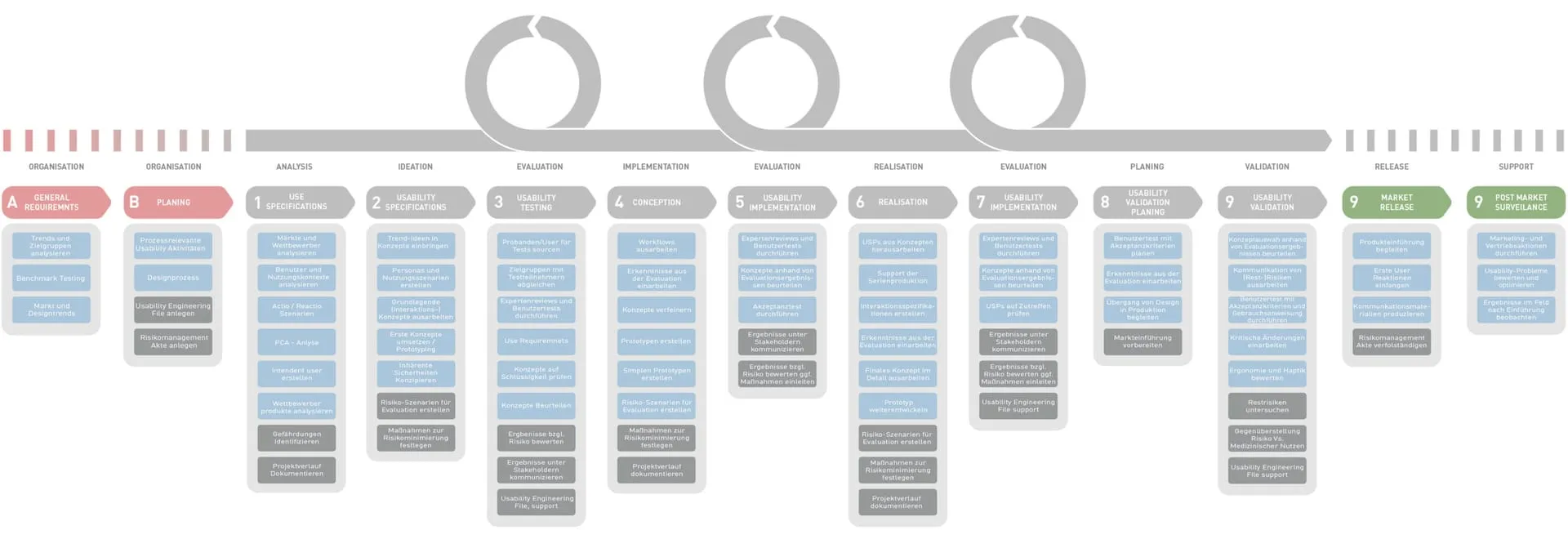
Der Medical-Design-Prozess von Entwurfreich basiert auf der IEC 62366 und wird durch praxisbewährte Meilensteine ergänzt. Jede Phase erzeugt dokumentierte Ergebnisse, die direkt in die technische Dokumentation einfließen. Die folgenden sieben Schritte führen ein Medizinprodukt von den ersten Anforderungen bis zur summativen Evaluation — und bringen regulatorische Konformität mit Designinnovation in Einklang.

Generelle Anforderungen — Planung
Die Planung hat einen zentralen Stellenwert und wird von unserem gesamten Team sorgfältig durchgeführt. Hier werden neben den Trend- und Designanalysen die ersten Usability-Aktivitäten für den gesamten Prozess geplant. Der Usability-Engineering-Plan definiert Umfang, Nutzergruppen, Evaluationsmethoden und Akzeptanzkriterien — das Fundament, auf dem jede weitere Phase aufbaut.
Use Specifications — Analyse
Zuerst wird der Nutzungskontext analysiert und Nutzer sowie Nutzungsgruppen (Intended User) erfasst. Durch verschiedene Analysemethoden, z. B. PCA-Analyse, können bereits potenzielle Interaktionsprobleme ermittelt werden. Parallel werden die Märkte und Wettbewerber untersucht, um dieses Wissen bereits in die Neuentwicklung einfließen zu lassen. Das Ergebnis ist eine dokumentierte Nutzungsspezifikation, die erfasst, wer das Gerät nutzt, wo, wie und warum.

Usability Specifications — Ideation
In diesem Schritt werden Nutzungsszenarien erstellt und erste grundlegende Potenziale für die Konzeption entwickelt. Ziel ist es, mögliche Use Errors zu entdecken und durch die Erarbeitung inhärenter Sicherheiten bereits frühzeitig vorzubeugen. Kritische Aufgaben werden definiert und priorisiert — das sind die Interaktionen, bei denen ein Anwendungsfehler den Patienten gefährden oder das Behandlungsergebnis beeinträchtigen könnte.
Usability Testing — Evaluation
Durch frühzeitige und iterative Testreihen auf formativem Level können erste Ergebnisse kostengünstig getestet werden. Mögliche Probleme werden so frühzeitig entdeckt und tragen zu einer wesentlichen Risikominimierung bei, indem sie bereits während der Entwicklung adressiert werden. Gängige Praxis und FDA-Leitlinien empfehlen mindestens 15 Teilnehmer pro Nutzergruppe für summative Evaluationen.


Usability Implementation — Realisation
Die aus den Konzepten erarbeiteten USPs werden verfeinert und Interaktionsspezifikationen erstellt. Prototypen werden weiterentwickelt und die Serienproduktion vorbereitet. Maßnahmen zur Risikominimierung werden festgelegt und umgesetzt. In dieser Phase werden Designentscheidungen gegen Fertigungsrestriktionen validiert — damit das finale Gerät nicht nur gebrauchstauglich, sondern auch produzierbar ist.
Summative Evaluation — Release
Hier erfolgt die Endkontrolle der Qualitätssicherung. Die Konzepte werden anhand von Evaluationsergebnissen beurteilt und Akzeptanztests durchgeführt. Die Prototypen sind seriennah ausgearbeitet und müssen die Akzeptanzkriterien erfüllen. Die summative Evaluation weist nach, dass nutzungsbezogene Risiken auf ein akzeptables Niveau reduziert wurden — ein obligatorisches Gate für die CE-Kennzeichnung.


Risikomanagement — Dokumentation
Das Risikomanagement ist systematisch in allen unseren Arbeitsschritten verankert. Ziel ist es, Risiken bereits während der Entwicklung zu reduzieren. Durch Risk Control Measures werden Use Errors frühzeitig erkannt und durch vorbeugende Maßnahmen abgeschätzt, bewertet und kontrolliert. Unsere Ergebnisse können direkt in die Risikomanagementakte eingearbeitet werden — das lebende Dokument, das das Gerät über seinen gesamten Lebenszyklus begleitet.
Unser detaillierter Prozess
Medical Design in der Praxis: Fallstudien
Kepler Dentalmikroskop — Ein chirurgisches Mikroskop für Zahnärzte und Oralchirurgen. Komplexes Artikulationssystem mit binokularer Optik und LED-Beleuchtung. Vollständiges IEC-62366-Usability-Engineering von der Nutzungskontextanalyse bis zur summativen Evaluation. Herausforderung: Präzisionsoptik und ergonomische Positionierung in beengten Behandlungsräumen in Einklang bringen.
Ondal VALIA Deckenpendel — Deckengetragener Pendelarm für Notaufnahmen und Intensivstationen. Modulares Design für krankenhausspezifische Konfigurationen. Interdisziplinäre Herausforderung: Das Gerät dient Pflegekräften, Anästhesisten und Gebäudemanagern mit grundlegend unterschiedlichen Anforderungen.
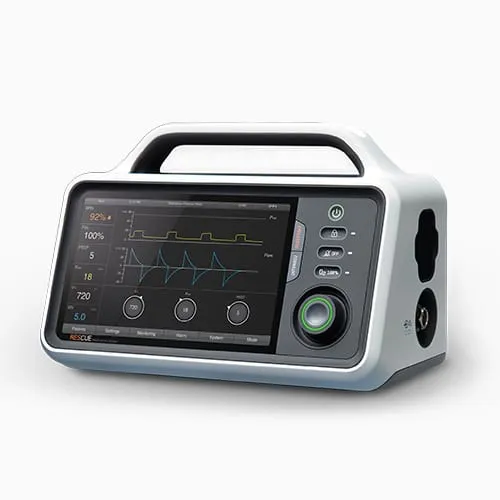
RESCUE Beatmungsgerät — Tragbare Beatmung für die Primär- und Sekundärversorgung. Touchscreen-Interface mit Echtzeit-Wellenformanzeige. Gestaltet für Rettungssanitäter, die unter Hochstress und eingeschränkter Sicht arbeiten — wo jede Sekunde Interaktionsverzögerung zählt.
Cactus Insulinmanagement — Ein täglicher Begleiter für Menschen mit Diabetes. Vernetztes Gerät mit E-Ink-Display und Smartphone-App. Red Dot Award 2014. Der Beweis, dass Medical Design sich persönlich statt klinisch anfühlen kann.
Raiing Temp+ Wearable — Gesundheits-Wearable mit 3-Elektroden-EKG, SpO2 und Temperaturüberwachung. Medizinische Genauigkeit in einem verbraucherfreundlichen Silikondesign. Zeigt, wie branchenübergreifendes Designdenken Unterhaltungselektronik und medizinische Regulierung verbindet.
Was kann eine Industriedesign-Agentur zum Medical Design beitragen?
Medical Design erfordert mehr als regulatorisches Wissen. Es erfordert Designdenken, das klinische Bedürfnisse, Fertigungsrestriktionen und Nutzerempathie verbindet. Genau hier schafft eine branchenübergreifende Industriedesign-Agentur einen Mehrwert, den reine MedTech-Beratungen nicht bieten können.
Ein Dentalmikroskop profitiert von derselben ergonomischen Sorgfalt, die bei Elektrowerkzeugen angewandt wird. Ein Beatmungsgeräte-Interface profitiert vom selben Interaktionsdesign wie Smart-Home-Steuerungen. Ein Insulinmanagement-Gerät profitiert vom selben Lifestyle-Produkt-Denken, das Unterhaltungselektronik formt. Branchenübergreifende Erfahrung verwässert medizinische Expertise nicht — sie verstärkt sie.
Als Industriedesign-Agentur hat Entwurfreich über 350 Projekte für 125+ Kunden realisiert, darunter ABB, Vodafone, Henkel, Coca-Cola, Fujifilm und Covestro. Das Team hat umfassende Erfahrung im Medical Design — vom Dentalmikroskop bis zum tragbaren Beatmungsgerät — alle entwickelt unter Einhaltung von IEC 62366 und MDR 2017/745/EU. Jüngste Auszeichnungen: iF Design Award Gold 2024, Red Dot Best of the Best 2024, German Design Award Gold 2026.
Ausgewählte Projekte


Häufig gestellte Fragen
Was ist Medical Design?
Medical Design ist die Disziplin der Entwicklung von Medizinprodukten, die sicher, wirksam, gebrauchstauglich und ästhetisch überzeugend sind. Sie integriert Usability Engineering (IEC 62366), Risikomanagement (ISO 14971) und regulatorische Konformität (MDR 2017/745/EU) mit Industriedesign-Methoden. Das Ziel sind Geräte, die die Zulassungsprüfung bestehen, in klinischen Umgebungen Vertrauen gewinnen und Marken in einem 586-Milliarden-Dollar-Markt differenzieren. Entwurfreich, eine Industriedesign-Agentur in Düsseldorf, wendet diese Disziplin über das gesamte Spektrum an — vom Dentalmikroskop und chirurgischer Beleuchtung bis zum Gesundheits-Wearable.
Was ist die IEC 62366 und warum ist sie wichtig?
Die IEC 62366 ist die internationale Norm für Usability Engineering von Medizinprodukten. Sie schreibt einen strukturierten Prozess vor: Nutzungskontextanalyse, Benutzerschnittstellenspezifikation, formative Evaluation und summative Evaluation. Benannte Stellen fordern dokumentierte Nachweise der IEC-62366-Konformität für die CE-Kennzeichnung unter MDR 2017/745/EU. Anwendungsfehler sind die häufigste Einzelursache für Medizinprodukte-Vorfälle — die IEC 62366 ist das Rahmenwerk, um sie zu verhindern. Die Norm gilt gleichermaßen für einfache Klasse-I-Geräte und komplexe Klasse-III-Systeme.
Was kostet Medical Design?
Die Kosten hängen von der Gerätekomplexität, Risikoklasse und dem regulatorischen Pfad ab. Einfache Klasse-I-Geräte erfordern weniger Dokumentation. Klasse IIb und III verlangen umfassendes Usability Engineering, klinische Bewertung und summative Prüfung. Eine Industriedesign-Agentur definiert den Umfang von Medical-Design-Projekten typischerweise von der Erstrecherche bis zu produktionsreifen Ergebnissen. Kontaktieren Sie uns für eine Projekteinschätzung.
Wie lange dauert die Entwicklung eines Medizinprodukts?
Die Zeitrahmen hängen von der Gerätekomplexität, Risikoklasse und dem regulatorischen Pfad ab. Ein einfaches Klasse-I-Gerät (z. B. ein chirurgisches Instrument) kann in 12 bis 18 Monaten vom Konzept zur CE-Kennzeichnung gelangen. Klasse IIa und IIb erfordern typischerweise 18 bis 30 Monate, einschließlich formativer und summativer Evaluationszyklen nach IEC 62366. Klasse-III-Geräte mit klinischen Prüfungen können 3 bis 5 Jahre dauern. Bei Entwurfreich umfassen die Industriedesign- und Usability-Engineering-Phasen — von der Erstrecherche bis zu produktionsreifen Ergebnissen — typischerweise 6 bis 14 Monate innerhalb des Gesamtentwicklungszeitraums.
Brauche ich die IEC 62366 für ein Klasse-I-Medizinprodukt?
Ja. Die IEC 62366 gilt für alle Medizinprodukte unabhängig von der Risikoklasse. Auch ein einfaches Klasse-I-Gerät muss nachweisen, dass nutzungsbezogene Risiken identifiziert und gemindert wurden. Umfang und Tiefe der Usability-Engineering-Datei skalieren mit der Gerätekomplexität — ein Klasse-I-Gerät erfordert weniger umfangreiche Dokumentation als ein Klasse-III-System —, aber der grundlegende Prozess (Nutzungskontextanalyse, Gefährdungsidentifikation, Evaluation) ist verpflichtend. Benannte Stellen und zuständige Behörden erwarten dokumentierte Nachweise der IEC-62366-Konformität für alle CE-gekennzeichneten Geräte unter MDR 2017/745/EU.
Wie misst man den Erfolg von Medical Design?
Der Erfolg von Medical Design wird über regulatorische und nutzerbezogene Metriken gemessen. Regulatorisch: CE-Kennzeichnung planmäßig erreicht, keine kritischen Befunde bei Audits durch benannte Stellen, vollständige Usability-Engineering-Datei akzeptiert. Nutzermetriken: Task-Completion-Rate bei summativer Evaluation (Ziel: >95 % bei kritischen Aufgaben), Fehlerrate-Reduktion im Vergleich zu Vorgängergeräten, System Usability Scale (SUS) über 68 (Branchendurchschnitt). Kommerzielle Metriken: Time-to-Market gegenüber Plan, Reduktion von Post-Market-Beschwerden, Net Promoter Score klinischer Nutzer. Am wirksamsten ist die Kombination aller drei Bereiche — regulatorische Konformität, Nutzerperformance und Geschäftsergebnisse.
Geschrieben von Simon Gorski · 15. Januar 2024