
A Strategic Guide for Product Managers, CTOs and Medical Device Manufacturers
Medical Design That Connects Regulation, Usability and Innovation
What Is Medical Design?
Medical design is the discipline of developing medical devices that are safe, effective and usable. Unlike consumer product design, every decision must satisfy a regulatory framework: MDR 2017/745/EU for European market access, IEC 62366 for usability engineering, ISO 14971 for risk management and ISO 13485 for quality management systems.
But compliance alone does not make a great product. The best medical design balances regulatory requirements with clinical workflows, user empathy and manufacturing feasibility. It is where engineering meets human factors — where the weight of a surgical instrument, the feedback of a ventilator button and the readability of an infusion pump display all carry clinical consequences.
For product managers and CTOs, medical design is not a downstream styling exercise. It is a strategic discipline that determines time-to-market, CE marking success, user adoption and long-term patient safety.
Why Does Medical Design Matter for Your Business?

The numbers speak clearly. The global medical devices market reached $586 billion in 2025 and is projected to grow to $1,084 billion by 2035 at a CAGR of 6.34% (source: Towards Healthcare). Medical device design and development services alone are valued at $13.29 billion, growing to $28.08 billion by 2034 at 8.6% CAGR (source: Fortune Business Insights).
Wearable medical devices will exceed $60 billion by 2026. Key trends for 2026 include AI-powered diagnostics, point-of-care testing, sustainable materials and robotic surgery. The EUDAMED database becomes mandatory in May 2026, adding another compliance layer.
According to Boehm's cost escalation model, late-stage usability problems cost 10 to 100 times more to fix than issues caught during early formative evaluation. For product managers, medical design is not a cost centre — it is the critical path to market access, competitive differentiation and patient safety.
What Regulatory Framework Governs Medical Design in Europe?
Three pillars define the regulatory landscape for medical device design in Europe. Understanding them is not optional — it is the prerequisite for CE marking.
What does MDR 2017/745/EU require from manufacturers?
The Medical Device Regulation (MDR 2017/745/EU) replaced the older Medical Device Directive (MDD) in May 2021. It introduced stricter classification rules, mandatory clinical evaluation for all risk classes, comprehensive technical documentation and continuous post-market surveillance. The regulation significantly impacts small and medium-sized manufacturers — many have postponed launches or left the EU market entirely. A reform proposal published in December 2025 aims to ease the burden on SMEs while maintaining safety standards. For designers, MDR means every material choice, interface decision and labelling detail must be documented and justified.
How does IEC 62366 structure the usability engineering process?
IEC 62366 is the international standard that defines how manufacturers must integrate usability engineering into medical device development. It requires context-of-use analysis, user interface specification, identification of use-related hazards and both formative and summative evaluation. Use errors are the single largest cause of medical device incidents — IEC 62366 exists to prevent them. Notified bodies expect documented evidence of IEC 62366 compliance for CE marking. The standard applies to every device, from a simple blood pressure cuff to a complex surgical robot. For medical design teams, it is not bureaucracy — it is the framework that keeps patients safe.
Why does ISO 14971 risk management matter for design?
ISO 14971 requires systematic identification, estimation and control of risks throughout the product lifecycle. For designers, this means every decision has a risk dimension: material selection, interface layout, physical ergonomics, cleaning procedures — all feed into the risk management file. Usability engineering under IEC 62366 and risk management under ISO 14971 are two sides of the same coin. A handle that is too smooth to grip securely in a wet operating room is not just a design flaw — it is a risk that must be identified, evaluated and controlled.

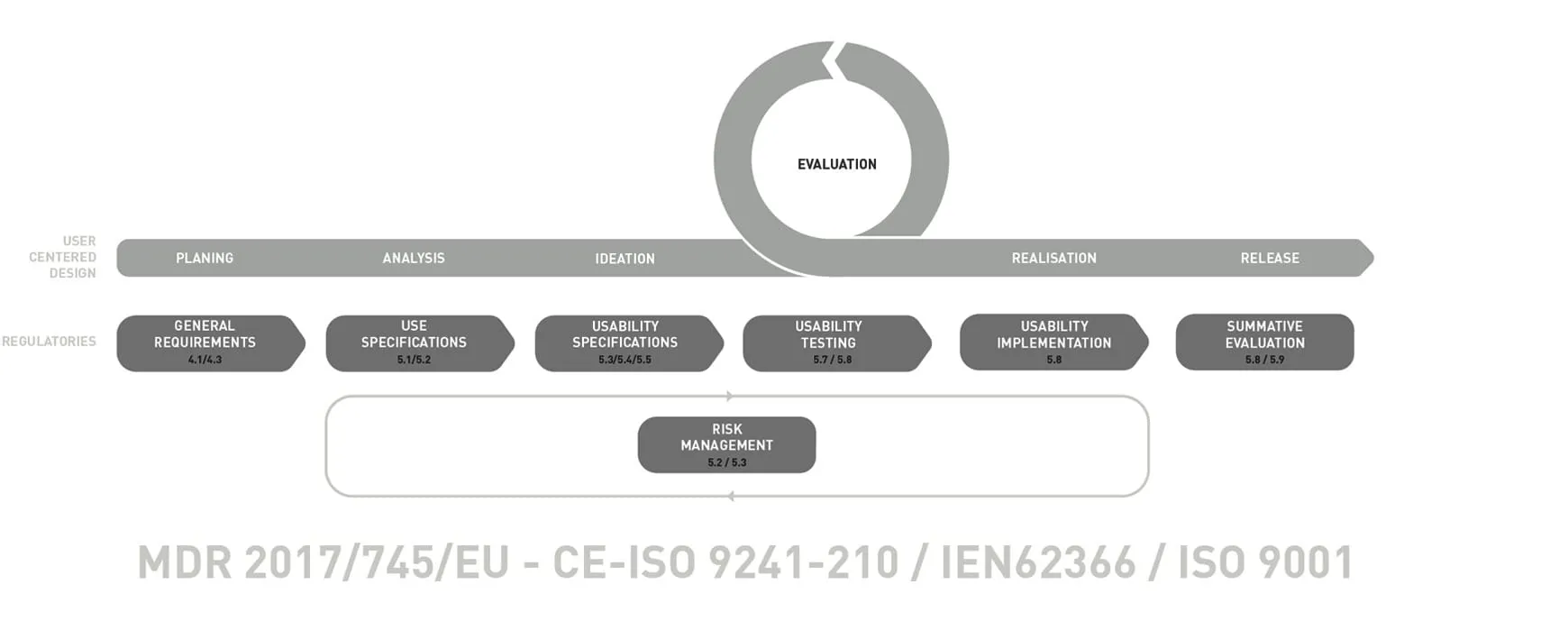
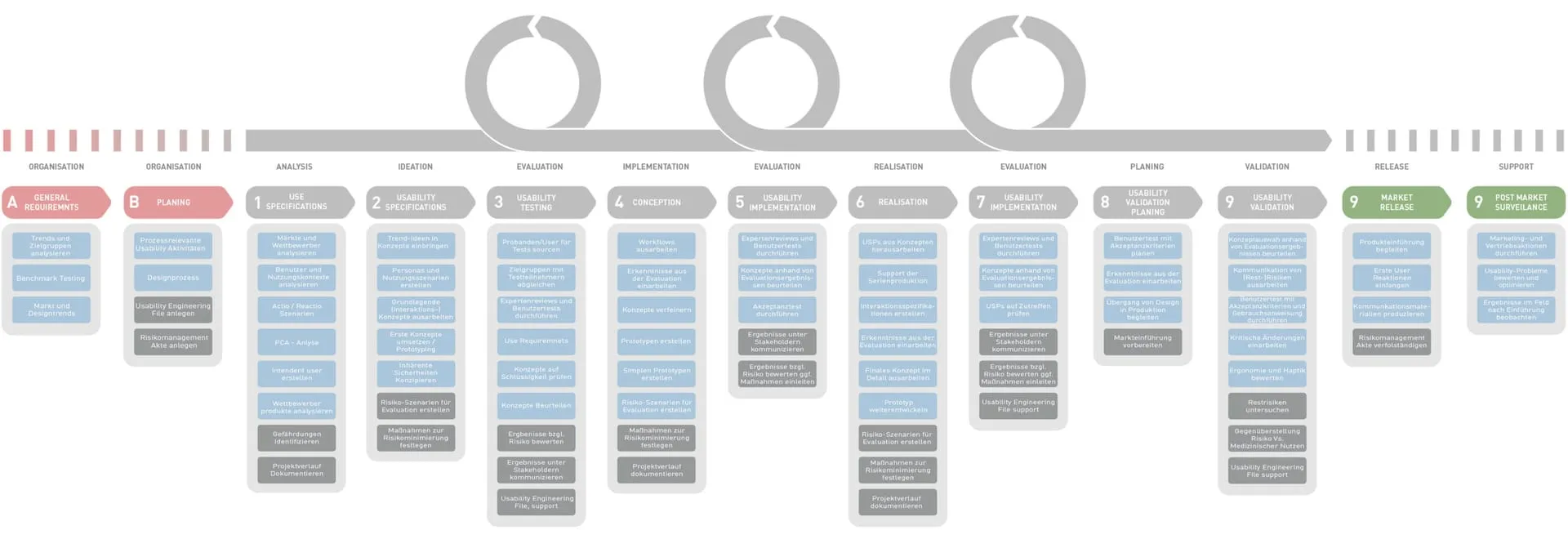
How Does a Seven-Step Usability Engineering Process Work?
Entwurfreich's medical design process is based on IEC 62366 and enhanced with proven milestones from practice. Each phase produces documented outputs that feed directly into the technical file. The following seven steps take a medical device from initial requirements to summative evaluation — balancing regulatory compliance with design innovation.

General Requirements — Planning
Planning holds a critical position in our process and is carried out meticulously by our entire team. In addition to trend and design analyses, the initial usability activities are mapped out for the whole project lifecycle. The usability engineering plan defines scope, user groups, evaluation methods and acceptance criteria — the foundation that every subsequent phase builds on.
Use Specifications — Analysis
First, the context of use is analysed and users and user groups (intended users) are identified. Through various analytical methods — such as PCA analysis — potential interaction problems can already be detected at this stage. In parallel, markets and competitors are examined so that these insights flow into the new development from the very beginning. The result is a documented use specification that captures who uses the device, where, how and why.

Usability Specifications — Ideation
In this step, usage scenarios are created and initial foundational concepts are developed. The goal is to identify potential use errors and to prevent them early on by establishing inherent safety measures. Critical tasks are defined and prioritised — these are the interactions where a use error could harm the patient or compromise treatment outcomes.
Usability Testing — Evaluation
Through early and iterative test cycles at a formative level, initial results can be tested cost-effectively. Potential problems are identified at an early stage, contributing significantly to risk reduction by addressing issues during development rather than after. Common practice and FDA guidance recommend a minimum of 15 participants per user group for summative evaluations.


Usability Implementation — Realisation
The USPs developed from the concepts are refined and interaction specifications are created. Prototypes are further developed and preparations for series production begin. Risk mitigation measures are defined and implemented. At this stage, design decisions are validated against manufacturing constraints — ensuring the final device is not only usable but also producible.
Summative Evaluation — Release
This is where the final quality assurance review takes place. Concepts are assessed based on evaluation results and acceptance tests are conducted. The prototypes are refined to near-production quality and must meet all acceptance criteria. The summative evaluation demonstrates that usability-related risks have been reduced to an acceptable level — a mandatory gate for CE marking.


Risk Management — Documentation
Risk management is systematically integrated into every step of our work. The goal is to reduce risks during development. Through risk control measures, use errors are identified early and assessed, evaluated and controlled through preventive actions. Our results can be directly incorporated into the risk management file — the living document that accompanies the device throughout its entire lifecycle.
Our Detailed Process
Medical Design in Practice: Real-World Case Studies
Kepler Dental Microscope — A surgical microscope designed for dentists and oral surgeons. Complex articulation system with binocular optics and LED illumination. Full IEC 62366 usability engineering from context-of-use analysis through summative evaluation. Challenge: balancing precision optics with ergonomic positioning in confined dental operatories.
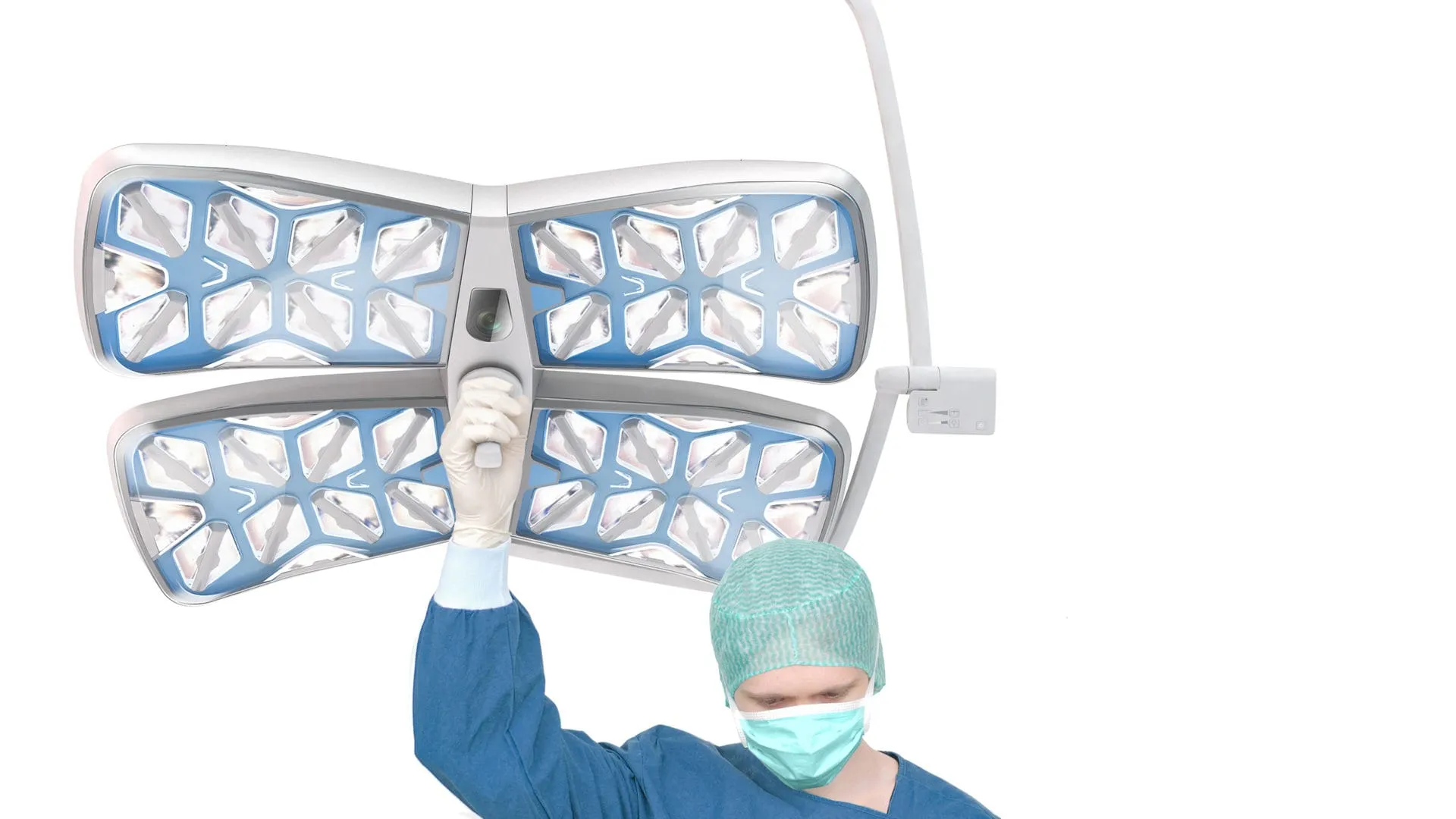
Ondal VALIA Pendant System — Ceiling-mounted medical pendant arm for emergency rooms and intensive care units. Modular design allowing hospital-specific configurations. Cross-disciplinary challenge: the device serves nurses, anaesthesiologists and facility managers with fundamentally different needs.
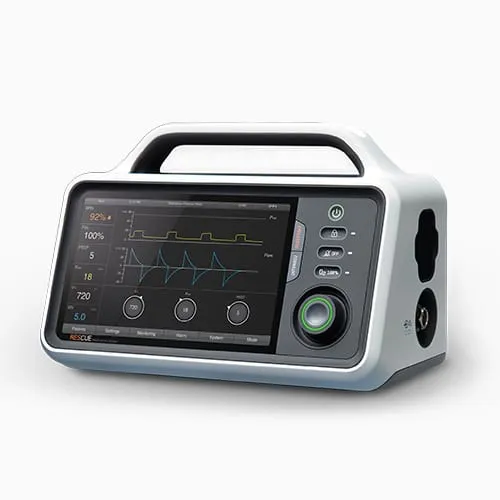
RESCUE Ventilator — Portable ventilation for primary and secondary care. Touchscreen interface with real-time waveform display. Designed for paramedics working in high-stress, low-visibility environments where every second of interaction delay matters.
Cactus Insulin Management — A daily companion for people with diabetes. Connected device with e-ink display and smartphone app. Red Dot Award 2014. Proof that medical design can feel personal rather than clinical.
Raiing Temp+ Wearable — Health care wearable with 3-electrode ECG, SpO2 and temperature monitoring. Medical-grade accuracy in a consumer-friendly silicone form factor. Demonstrates how cross-sector design thinking bridges consumer electronics and medical regulation.
What Can an Industrial Design Agency Contribute to Medical Design?
Medical design requires more than regulatory knowledge. It requires design thinking that bridges clinical needs, manufacturing constraints and user empathy. This is where a cross-sector industrial design agency adds value that pure MedTech consultancies cannot.
A dental microscope benefits from the same ergonomic rigour applied to power tools. A ventilator interface benefits from the same interaction design used in smart home controls. An insulin management device benefits from the same lifestyle-product thinking that shapes consumer electronics. Cross-sector experience does not dilute medical expertise — it amplifies it.
As an industrial design agency, Entwurfreich has completed over 350 projects for 125+ clients including ABB, Vodafone, Henkel, Coca-Cola, Fujifilm and Covestro. The team has extensive experience in medical device design — from dental microscopes to portable ventilators — all developed under IEC 62366 and MDR 2017/745/EU compliance. Recent awards: iF Design Award Gold 2024, Red Dot Best of the Best 2024, German Design Award Gold 2026.
Selected Projects


Frequently Asked Questions
What is medical design?
Medical design is the discipline of developing medical devices that are safe, effective, usable and aesthetically compelling. It integrates usability engineering (IEC 62366), risk management (ISO 14971) and regulatory compliance (MDR 2017/745/EU) with industrial design methods. The goal is devices that pass regulatory review, win user trust in clinical environments and differentiate brands in a $586 billion global market. Entwurfreich, an industrial design agency in Dusseldorf, applies this discipline across the full spectrum — from dental microscopes and surgical lighting to wearable health monitors.
What is IEC 62366 and why does it matter?
IEC 62366 is the international standard for usability engineering of medical devices. It mandates a structured process: context-of-use analysis, user interface specification, formative evaluation and summative evaluation. Notified bodies require documented evidence of IEC 62366 compliance for CE marking under MDR 2017/745/EU. Use errors are the single largest cause of medical device incidents — IEC 62366 is the framework for preventing them. The standard applies equally to simple Class I devices and complex Class III systems.
How much does medical device design cost?
Cost depends on device complexity, risk class and regulatory pathway. Simple Class I devices require less documentation. Class IIb and III devices demand extensive usability engineering, clinical evaluation and summative testing. An industrial design agency typically scopes medical design projects from initial research through production-ready deliverables. Contact us for a project estimate.
How long does medical device development take?
Timelines depend on device complexity, risk class and regulatory pathway. A simple Class I device (e.g. a surgical instrument) can move from concept to CE marking in 12 to 18 months. Class IIa and IIb devices typically require 18 to 30 months, including formative and summative evaluation cycles under IEC 62366. Class III devices with clinical investigations can take 3 to 5 years. At Entwurfreich, the industrial design and usability engineering phases — from initial research through production-ready deliverables — typically span 6 to 14 months within the overall development timeline.
Do I need IEC 62366 for a Class I medical device?
Yes. IEC 62366 applies to all medical devices regardless of risk class. Even a simple Class I device must demonstrate that usability-related risks have been identified and mitigated. The scope and depth of the usability engineering file scales with device complexity — a Class I device requires less extensive documentation than a Class III system — but the fundamental process (context-of-use analysis, hazard identification, evaluation) is mandatory. Notified bodies and competent authorities expect documented evidence of IEC 62366 compliance for all CE-marked devices under MDR 2017/745/EU.
How do you measure the success of medical design?
Medical design success is measured through regulatory and user metrics. Regulatory: CE marking achieved on schedule, no critical findings from notified body audits, complete usability engineering file accepted. User metrics: task completion rate during summative evaluation (target: >95% for critical tasks), error rate reduction compared to predicate devices, System Usability Scale (SUS) scores above 68 (industry average). Commercial metrics: time-to-market versus plan, reduction in post-market complaints, Net Promoter Score from clinical users. The most effective approach combines all three — regulatory compliance, user performance and business outcomes.
Written by Simon Gorski · January 15, 2024